
To streamline the procedure, MIT scientists produced a machine-learning design that can directly forecast the complex that will form when 2 proteins bind together. Their method is in between 80 and 500 times faster than cutting edge software approaches, and frequently anticipates protein structures that are closer to actual structures that have been observed experimentally.
This technique could assist researchers better understand some biological procedures that involve protein interactions, like DNA replication and repair work; it might likewise accelerate the process of developing brand-new medications.
” Deep knowing is really excellent at capturing interactions in between different proteins that are otherwise hard for chemists or biologists to write experimentally. A few of these interactions are very made complex, and individuals have not found excellent methods to reveal them. This deep-learning model can discover these kinds of interactions from information,” says Octavian-Eugen Ganea, a postdoc in the MIT Computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead author of the paper.
Ganeas co-lead author is Xinyuan Huang, a college student at ETH Zurich. MIT co-authors include Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in CSAIL, and Tommi Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Data, Systems, and Society. The research study will be provided at the International Conference on Learning Representations.
Protein attachment
The model the researchers developed, called Equidock, focuses on stiff body docking– which takes place when two proteins connect by translating or turning in 3D space, however their shapes do not squeeze or bend.
The design takes the 3D structures of 2 proteins and converts those structures into 3D charts that can be processed by the neural network. Proteins are formed from chains of amino acids, and each of those amino acids is represented by a node in the chart.
The scientists included geometric understanding into the model, so it comprehends how items can alter if they are turned or equated in 3D area. The model also has actually mathematical knowledge built in that makes sure the proteins constantly attach in the exact same way, no matter where they exist in 3D space. This is how proteins dock in the body.
Using this details, the machine-learning system identifies atoms of the 2 proteins that are most likely to communicate and form chain reactions, referred to as binding-pocket points. It uses these points to place the 2 proteins together into a complex.
” If we can comprehend from the proteins which private parts are likely to be these binding pocket points, then that will capture all the details we need to position the 2 proteins together. Assuming we can discover these 2 sets of points, then we can simply find out how to rotate and translate the proteins so one set matches the other set,” Ganea explains.
Among the most significant challenges of building this model was overcoming the lack of training data. Since so little experimental 3D information for proteins exist, it was particularly essential to incorporate geometric knowledge into Equidock, Ganea says. Without those geometric constraints, the design may get incorrect connections in the dataset.
Hours vs. seconds
Once the design was trained, the scientists compared it to four software approaches. Equidock is able to forecast the last protein complex after just one to five seconds. All the baselines took much longer, from between 10 minutes to an hour or more.
In quality procedures, which determine how carefully the predicted protein complex matches the real protein complex, Equidock was typically similar with the standards, but it in some cases underperformed them.
” We are still lagging behind one of the standards. Our approach can still be improved, and it can still work. It might be used in a large virtual screening where we wish to comprehend how thousands of proteins can engage and form complexes. Our technique could be utilized to generate an initial set of candidates extremely quick, and after that these might be fine-tuned with a few of the more precise, however slower, standard methods,” he states.
In addition to utilizing this technique with standard designs, the team wishes to include specific atomic interactions into Equidock so it can make more accurate predictions. In some cases atoms in proteins will attach through hydrophobic interactions, which involve water molecules.
Their strategy could likewise be used to the advancement of small, drug-like particles, Ganea says. These particles bind with protein surfaces in particular ways, so quickly identifying how that attachment takes place could reduce the drug advancement timeline.
In the future, they plan to improve Equidock so it can make forecasts for flexible protein docking. The most significant difficulty there is an absence of information for training, so Ganea and his coworkers are working to generate synthetic data they could utilize to enhance the design.
Reference: “Independent SE( 3 )- Equivariant Models for End-to-End Rigid Protein Docking” by Octavian-Eugen Ganea, Xinyuan Huang, Charlotte Bunne, Yatao Bian, Regina Barzilay, Tommi S. Jaakkola and Andreas Krause, 28 September 2021, ICLR 2022 Conference.OpenReview
This work was moneyed, in part, by the Machine Learning for Pharmaceutical Discovery and Synthesis consortium, the Swiss National Science Foundation, the Abdul Latif Jameel Clinic for Machine Learning in Health, the DTRA Discovery of Medical Countermeasures Against New and Emerging (DOMANE) threats program, and the DARPA Accelerated Molecular Discovery program.
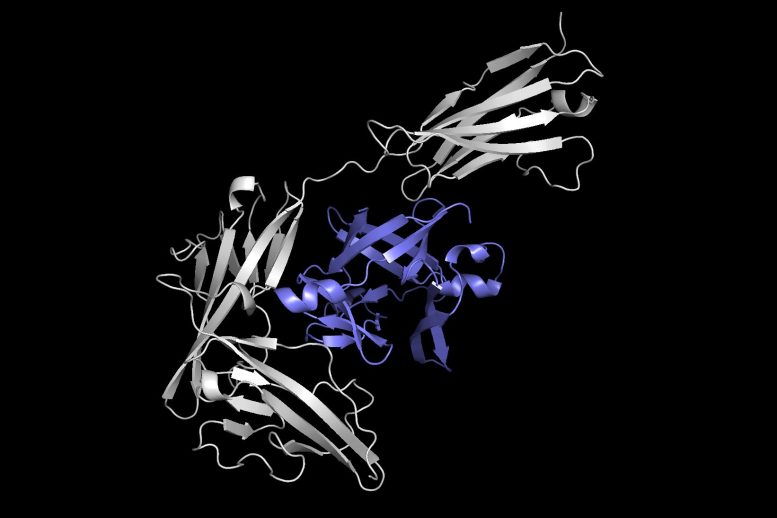
This image reveals one protein (in gray) docking with another protein (in purple) to form a protein complex. Equidock, the machine knowing system the scientists developed, can straight anticipate a protein complex like this in a matter of seconds. The model also has mathematical knowledge constructed in that makes sure the proteins constantly attach in the exact same way, no matter where they exist in 3D space. Equidock is able to anticipate the final protein complex after just one to five seconds. It might be used in a very large virtual screening where we desire to understand how thousands of proteins can engage and form complexes.
This image reveals one protein (in gray) docking with another protein (in purple) to form a protein complex. Equidock, the artificial intelligence system the scientists established, can directly forecast a protein complex like this in a matter of seconds. Credit: Courtesy of the scientists
The machine-learning model might assist scientists speed the advancement of new medications.
Antibodies, small proteins produced by the immune system, can connect to particular parts of a virus to neutralize it. As scientists continue to battle SARS-CoV-2, the infection that causes Covid-19, one possible weapon is an artificial antibody that binds with the infection spike proteins to avoid the infection from entering a human cell.
To develop a successful artificial antibody, scientists need to understand precisely how that attachment will occur. Proteins, with bumpy 3D structures including lots of folds, can stick in countless combinations, so finding the ideal protein complex among almost numerous prospects is very time-consuming.